Gromacs is a free package designed for the simulation of proteins, nucleic acid and small molecules. It is easy for beginners compared to other simulation packages. For best performance, you may want to install it on a cluster, but here I just want to show what a local laptop can do, so the method here may differ from that of cluster installation.

Image from here
Step A. Download and install Gromacs.
If you are a chemistry or biology guy, you probably not a fan of coding, there are quick installation lines for you. The installation need “cmake”, I recommend you just go ahead with the following lines, if you are required to install CMake, then just install it, if nothing wrong means your system already contains CMake, you don’t need to worry about it then.
The quick installation need you to put the lines below in your terminal, after you have downloaded the Gromacs 2020.targz, from Gromacs offical downloading site This is a "dirty" installation so there is no speed optimisation based on your computer, sadly as its offical documentation mentioned, if you do want to make a "perfect" installation to make full use of your computer, you have to read a lot more techinical stuff, for which I try to avoid here
There are 9 commonds in total which I have give each of them a number, I will use these nummber in the installation process
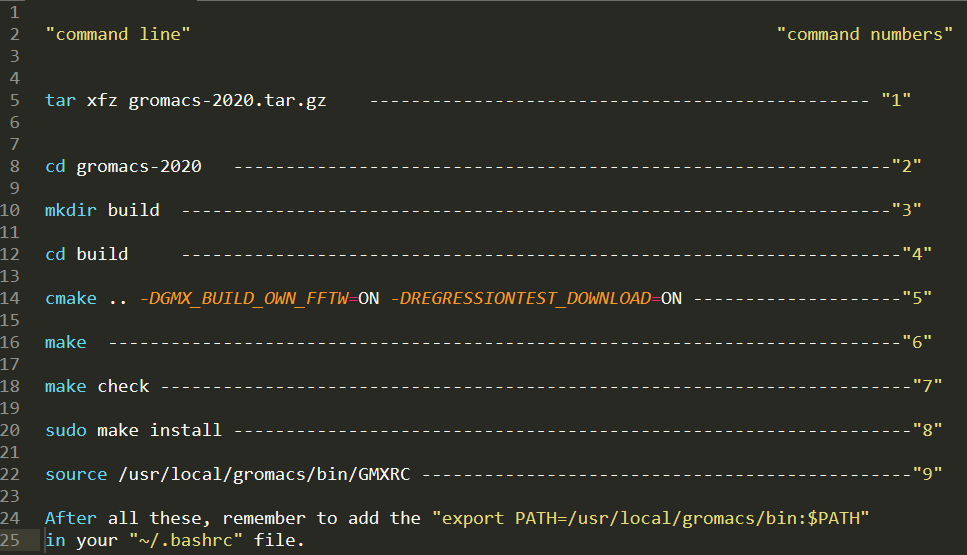
command "1" is just try to unzip to extract the file to a folder called "gromacs-2020" Comand "2" is entering the newly extracted folder, and "3" mean to creat a "build" folder inside "gromacs-2020". "4" let you enter to build, and prepare to run "5", the output of "5" will be stored in "build"
Input command "5", wait until finish
There might be a warning saying things about GPU, just ignore it and move on
Input command "6", after finish, it looks like below
Command "6" may take a while before finish, after finish, it looks like
Input comand "7", after finish, it looks like below
Input comand "8", after finish, it looks like below
Input comand "9", hit enter button
last, Open the ~/.bashrc
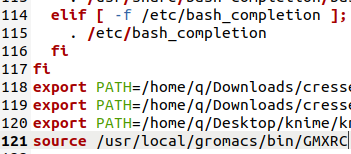
Add either "source /usr/locall/gromacs/bin/GMXC"
or "export PATH=/usr/locall/gromacs/bin:$PATH"
to the last line of your ~/.bashrc file.Try “gmx -h”, press enter, if you can find a lovely "Gromacs reminds you:" like in the green box, congratulations, you already get Gromacs installed on your laptop
Step B. Head to This Tutorial ,do the simulation.
This a very nice and concise tutorial you can follow step by step, so I will not go to the details too much. After you download the protein from PDB bank, you can almost copy the command lines from the "Lysozyme in water" tutorial, and hit enter to run your simulation.
But just be aware all the *.mdp files need YOU to creat them,. you should pay attention to the RED content like "here" or "this" when you go throung the "Lysozyme in water" tutorial, copy the content from the RED links, and paste to your *.mdp file
Step C. Head to Output analysis ,do more analysis.
In the "Lysozyme in Water" tutorial, you probably already did several analysis, but it may not enough, so I recommend you to use this site to do more analysis like RMSF and H bond networks analysis.
Please beaware, you can choose other analysis means, but for the sake of this blog, it is good to use the article was put here, it is consistent with the "Lysozyme in water" tutorial, so you can just copy the command line from this site to paste directly to your terminal and run it, just like what you have done in Step B
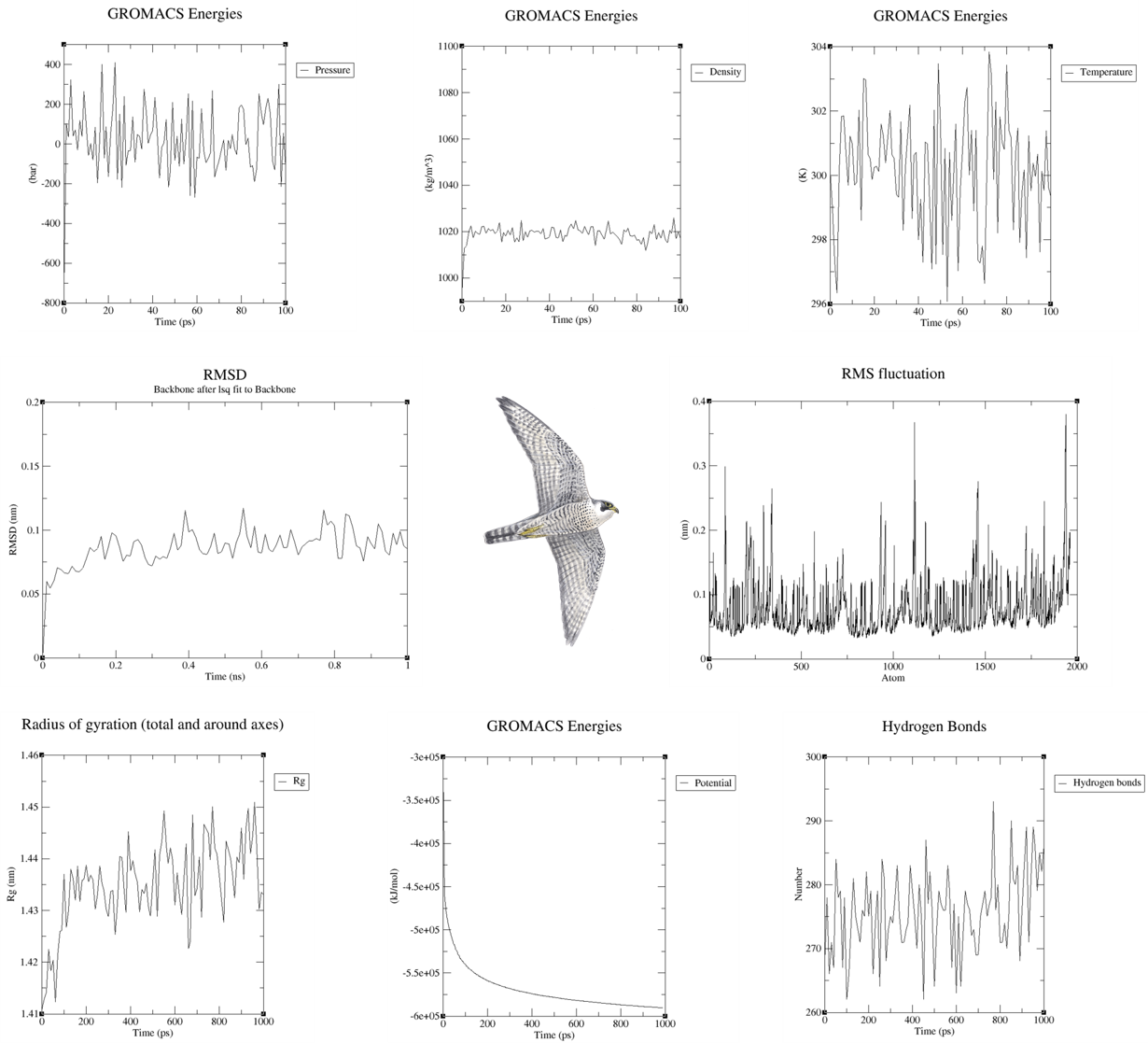
A total of 8 plots were obtained including the RMSD plot, by which we can tell the protein in water kind of reached equilibrium, with little fluctuations. RMSD plot usually tells you if your simulation is good or not, also, some more complex plot like the hydrogen bonding networks between "non-protein" and protein were created as well, in this case, there is no small ligand but a pure protein, so the "non-protein" means the environment like waters and ions
By finishing this, you can get a sense of what Gromacs can do, of course, there is a lot more Gromacs can do, like predicting the protein folding process by a very long time simulation which can only be done by a cluster or supercomputer.
Further Reading
As we mentioned, this blog uses a pure protein without ligand for the sake of saving simulation time and make things easier for learning purposes. More than often, we are interested in a protein-ligand complex, in that case, after finishing the simulation, if we plot out the H-bonding networks between the ligand and protein, the information will be valuable to judge if a certain H-bond is strong or weak, which can help drug-like molecule design.
What's more, there are some excellent implementations you could use like the MMGBSA analysis, which can help to determine how much a certain pocket amino acid contribute to the ligand bind, which may be helpful when you try to mutant a key amino acid.